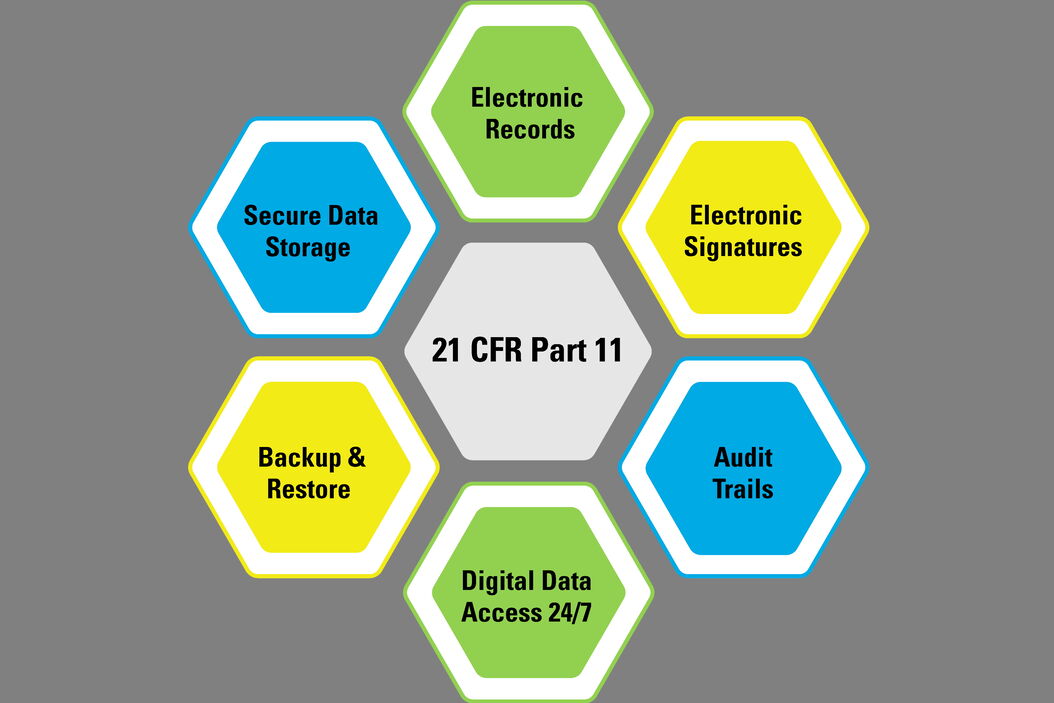
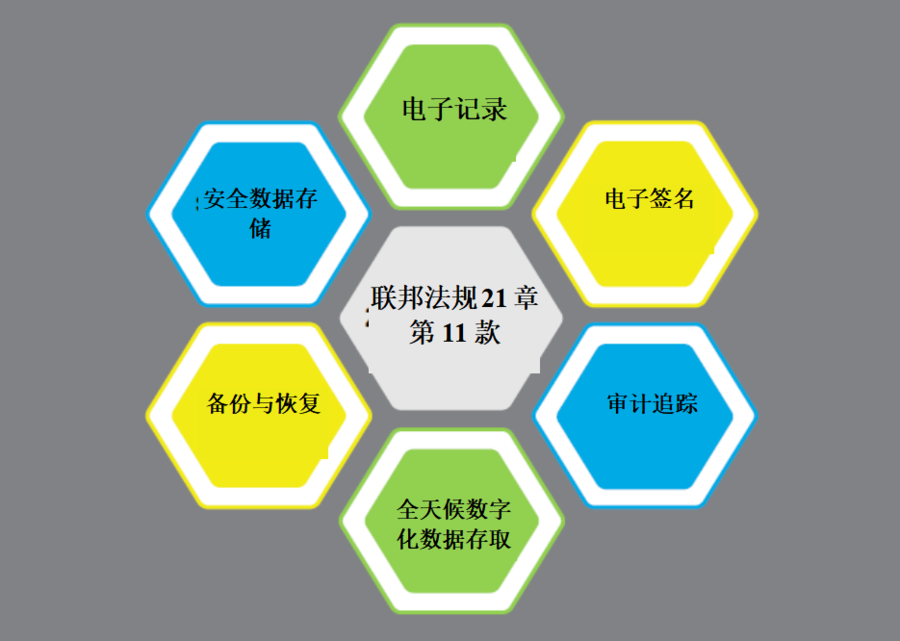
现有的数字化检验方法和满足电子记录相关法规的要求
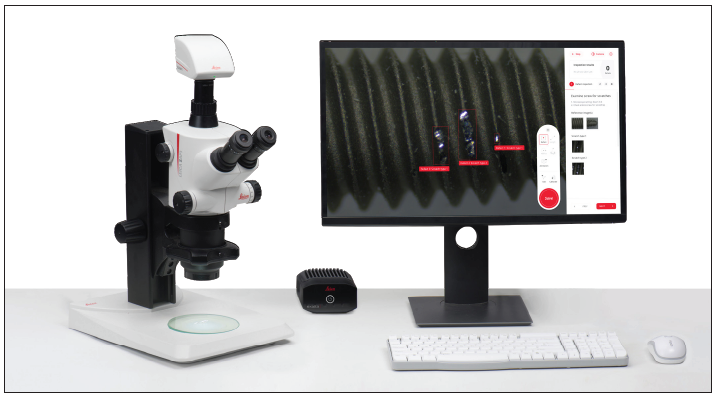
目前用于医疗器械的目测检查方法还没有提高整体检查过程实用性的系统和软件,即有许多手动步骤来检查器械以及输入和传输数据[4,5] 。因此,这会降低满足有关电子记录、批准和签名的法规和指南要求的效率。Leica Microsystems生产的用于可追溯性显微镜的Exalta智能设备(参见图1),现已上市,可帮助用户:i)为医疗器械的实际显微镜检查建立数字化增强解决方案,尽可能地减少手动步骤[4,5] ;ii)由于易于记录可追溯性、电子签名和批准、快速的用户身份验证和实用的审计追踪功能,为符合法规要求做好准备,例如联邦法规第21章第11款和GMP附录11等法规。
总结
本报告概述了美国(FDA联邦法规第21章第11款)、欧盟(GMP附录11)和中国(NMPA规范)关于医疗器械检查中使用电子记录的法规。数字化增强检查比纸质记录方法有显著的优势。这些优势可以产生更一致和更有效的检查结果。但是,电子记录的规定与纸质记录的要求不同,并且可能因国家而异。目前,制造商使用的目测检查方法可能有许多手动步骤,从而降低了遵循这些电子记录规定的效率。Leica Microsystems生产的用于可追溯性显微镜的Exalta智能设备使用户能够建立数字增强化的检查解决方案,以实现更有效的医疗器械质量控制,并帮助他们为符合此类电子记录法规的要求做好准备。
相关文章
-

验证汽车零部件的规格
在汽车零部件的开发和生产过程中,无论是供应商还是汽车制造商,都必须符合规格要求。这些规格对保持汽车和其他车辆在生命周期内的性能标准和安全运行至关重要[1,2,3]。在满足或超越日益严格的质量标准的同时…
Feb 20, 2025Read article -

晶圆上的光刻胶残留和有机污染物的可视化
随着半导体上集成电路(IC)的尺寸低于10纳米,在晶圆检测中有效检测光刻胶残留等有机污染物和缺陷变得越来越重要。光学显微镜仍然是常见的检测方法,但对于有机污染物,明场和其他类型的照明可能会存在局限性。…
Jun 12, 2024Read article -
 at the edge of a battery electrode acquired with a DVM6 digital microscope.")
电池制造过程中的毛刺检测
毛刺是电池电极片边缘可能出现的缺陷,例如在制造过程中的分切环节。它们可能会因诸如短路等故障导致电池性能下降,并引发安全和可靠性问题。毛刺检测是电池生产质量控制的重要部分,对于生产具有可靠性能和寿命的电…
Apr 04, 2024Read article
